
This blog post is provided by Nathaniel Mull and tells the #StoryBehindThePaper for the paper “Virus isolation data improve host predictions for New World rodent orthohantaviruses“, which was recently published in the Journal of Animal Ecology.
Nathaniel is a PhD student in the Fayetteville Disease Ecology lab at University of Arkansas. His dissertation is broadly examining how habitat management and community interactions influence the infection dynamics of wildlife pathogens, with a focus on the rodent-orthohantavirus system.
Most emerging infectious diseases (EIDs) in humans are caused by exposure to pathogens that naturally circulate in wildlife (zoonoses), and most cases are the result of unique exposure events. Although some EIDs are recurringly caused by interactions with wildlife known to host particular pathogens, many are the result of infection by unknown pathogens, often carried by species not known to host any pathogens that threaten human health. Therefore, the first step in mitigating risk of human exposure to EIDs is determining the breadth of pathogens that are present in the landscape and which species carry and transmit them.
Broad surveying of wildlife is resource-intensive, requiring extensive sampling of diverse taxa over wide spatial and temporal scales. Novel pathogen discovery can be made more efficient by focusing efforts on a determined short-list of probable hosts. Because host species are often related and have similar life history traits, unknown hosts can be predicted by collating information on known hosts and applying it to potential host species.
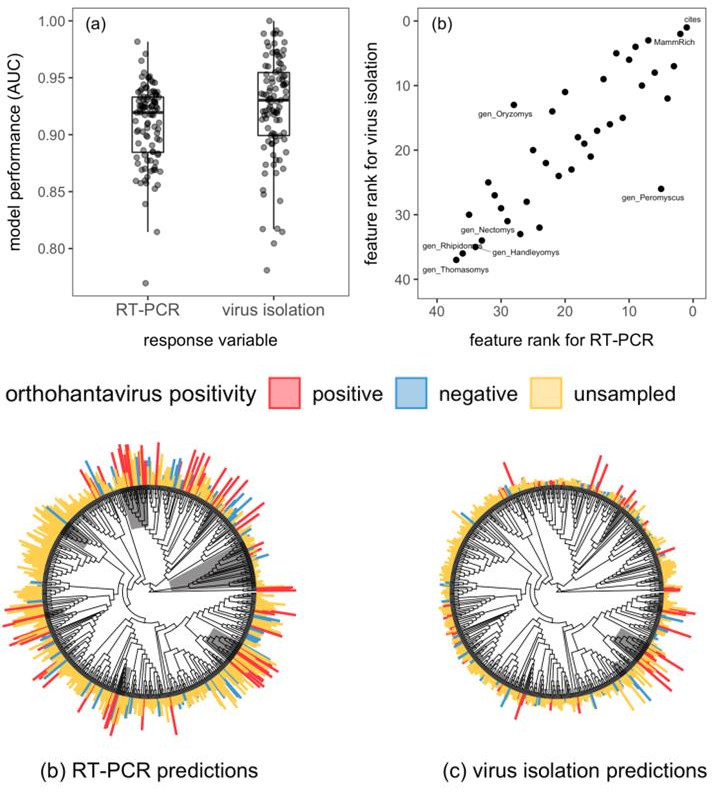
In our study recently published in Journal of Animal Ecology, we demonstrated the similarities and differences between predictive models using different levels of evidence of being a host (RT-PCR versus virus isolation) using the rodent-orthohantavirus system. RT-PCR data demonstrate an active infection by a pathogen (host) but do not indicate the role or competence of the host in transmitting the pathogen to other individuals (reservoir host); virus isolation is the gold standard for identifying a species as a reservoir but is often difficult to perform due to low success rates. This analysis expanded on a previous review by several of the authors highlighting rapid discovery of and lack of research on orthohantaviruses in North and South America using a predictive modeling approach designed by the other authors for betacoronaviruses.
Using data collected from previous empirical studies, we identified and ranked rodent traits that are associated with orthohantavirus infection (RT-PCR) and competence (virus isolation) for use in predictive modeling. A similar set of traits were identified as predictive of positivity between data types, with number of citations, mammal species richness, and litter size ranking in the top 5 traits for both profiles. The trait profiles were applied to rodents without orthohantavirus host evidence to predict species that are likely to be unidentified orthohantavirus hosts. Although there was some overlap in host predictions (27 species), overall the predicted hosts differed substantially between data types, with the virus isolation model creating a more succinct list of predicted species (92 total species for virus isolation; 138 total species for RT-PCR). Notably, RT-PCR data was previously collected from several of the species predicted by the virus isolation model, validating the accuracy of this approach.

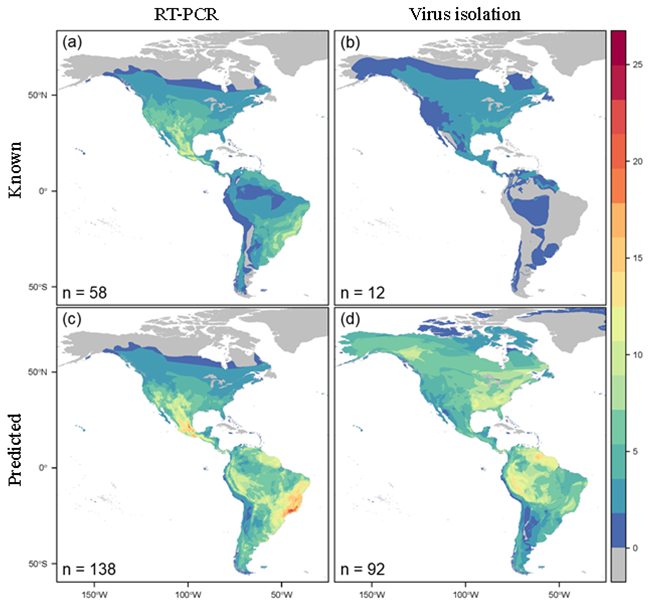
By overlapping predicted host distributions, we were able to identify hotspots of unknown hosts. Despite the large similarities in model performance, host distribution maps revealed stark differences in regional predictions. The distribution of hosts predicted by the RT-PCR model largely mirrored the distribution of known hosts based on RT-PCR data, while the distribution of hosts predicted by the virus isolation model varied greatly from the distribution of known hosts based on virus isolation data. These regions represent hotspots for future surveillance. In particular, the portions of Peru and northern South America surrounding the Amazon basin had the most overlap between distribution maps.
Interestingly, many of the regions with high predicted host density correspond with gaps in known orthohantavirus occurrence, including southern South America, the area surrounding the Amazon basin, southern Mexico, eastern United States and Canada, and northwestern Canada. Verifying orthohantavirus hosts and discovering novel orthohantaviruses that they likely carry will therefore provide information to not only mitigate human exposure, but also gain insight into how orthohantaviruses spread across the landscape. These insights would have been lost using a single type of evidence, highlighting the importance of our dual-model approach.
Our study examined American orthohantaviruses, but these methods are transferable to other virus systems and potentially other non-virus pathogen systems. For example, although many Eurasian orthohantavirus-host systems are well-established, neighboring regions such as the Middle East and Africa would clearly benefit from our model system. Predictive modeling would be useful for other virus groups as well, such as orthopoxviruses, which are commonly found in select taxa despite host roles being largely unknown. However, it’s important to note that this approach is limited to systems with enough prior evidence to properly train the models.
Although field work is necessary to validate our approach, the integration of multiple types of evidence enhances and hones predictive capabilities by combining the strengths and minimizing the weaknesses of each individual evidence type. The increase in long-term efficiency of focused surveillance will reduce resource costs, and innovative methodologies such as the strategy described in this article ultimately benefit human health by mitigating exposure risk to novel pathogens.
Read the paper
Read the full paper here: Mull, N., Carlson, C. J., Forbes, K. M. & Becker, D. J. (2022). Virus isolation data improve host predictions for New World rodent orthohantaviruses. Journal of Animal Ecology, 00, 1– 13. https://doi.org/10.1111/1365-2656.13694
